Disease Information
Summary
Resource(s) for Medical Professionals and Scientists on This Disease:
- GeneReview provides clinical information on genetic diseases, including diagnosis, treatment, and genetic counseling.
About Gaucher disease perinatal lethal
Many rare diseases have limited information. Currently, GARD aims to provide the following information for this disease:
- Symptoms:May start to appear as a Newborn and as an Infant.
- Cause:This disease has more than one possible cause.
- Organizations:Patient organizations dedicated to this rare disease are available on GARD, or you may contact a GARD Information Specialist for additional information.
- Categories:Genetic diseases
Skin diseases Inherited Metabolic diseases Lysosomal Storage diseases
Causes
What Causes This Disease?
Genetic Mutations
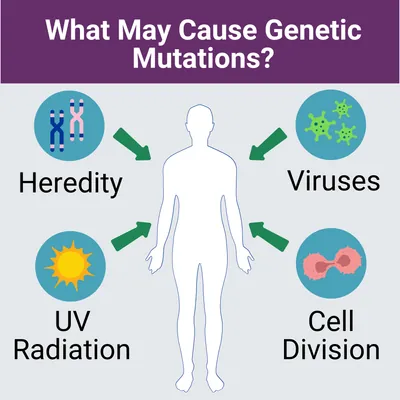
If you suspect you may have this disease, you may want to start collecting your family health history. Information such as other family members who have had similar symptoms, when their/your symptoms first appeared, or exposures to any potential disease-causing environmental factors should be discussed with your medical team. This tool from the Surgeon General can help you collect your family health history.
Can diseases be passed down from parent to child?
A biological parent can sometimes pass down genetic changes, called mutations, that cause a disease or increase the chances of developing it. This is called inheritance. Knowing if other family members have had the disease, also known as your family health history, can give your medical team important information. The Surgeon General offers a tool to help you collect your family health history.
Autosomal Recessive
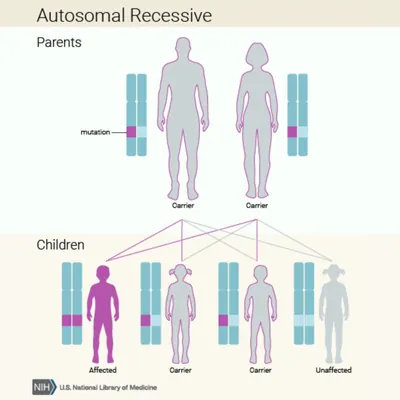
If both biological parents are carriers, there is a 25% their child inherits both copies of the mutated gene and is affected by the disease. Additionally, there is a 50% chance their child inherits only one copy of the mutated gene and is a carrier.
Learn more about inheritance patterns from the National Library of Medicine (NLM).
When Do Symptoms of Gaucher disease perinatal lethal Begin?
The age symptoms may begin to appear differs between diseases. Symptoms may begin in a single age range, or during several age ranges. The symptoms of some diseases may begin at any age. Knowing when symptoms may have appeared can help medical providers find the correct diagnosis.
Symptoms
The following describes the symptom(s) associated with this disease along with the corresponding body system(s), description, synonyms, and frequency (Note: Not all possible symptoms may be listed):
25 Symptoms
25 Symptoms
Musculoskeletal System
Medical Term
Arthrogryposis multiplex congenita
Navigating Health Care Decisions
On average, it can take more than six years to receive an accurate diagnosis. Many primary care providers (PCPs) may not be familiar with rare diseases, and patients often need to visit multiple specialists or seek second opinions to get answers.
If a diagnosis remains unclear, visiting a multidisciplinary care center or university hospital may help. These centers bring together teams of specialists who can work together to evaluate symptoms and coordinate a diagnosis. This team-based approach is also helpful after a diagnosis, when managing care for rare diseases.
Because only about 5% of rare diseases have FDA-approved treatments, finding the right healthcare team to manage your symptoms and overall health is essential. People living with rare diseases often face challenges such as delayed diagnosis, limited treatment options, and difficulty accessing knowledgeable providers. Building a care team that understands your needs can make a significant difference in your quality of life.

Your Health Care Team
Why is building the right health care team important?
Building the right health care team is key to the diagnosis, treatment, and management of your long-term health journey living with a rare disease. Start by choosing a primary care provider (PCP). Your PCP will be your main point of contact and help coordinate care with other medical professionals. Your PCP may order tests or refer you to specialists. To find a PCP near you, use the Medicare provider search tool and enter your location and “Primary Care Provider.”
Seeing multiple specialists is important for people with rare diseases because these conditions often affect many parts of the body and require care from doctors with different expertise. Most primary care providers may not be familiar with rare diseases, so involving specialists can lead to a more accurate diagnosis and better care. A coordinated team approach ensures that all symptoms are addressed and that care is well-managed. It can also connect patients with the latest research or treatment options.

You may want to consider bringing a copy of your medical history and a list of any medications to your appointment.
Ask your primary care provider for a referral or use this directory provided by the American College of Medical Genetics and Genomics to find a specialist.
You may want to consider bringing a copy of your medical history and a list of any medications to your appointment.
Ask your primary care provider for a referral or use this directory provided by the American Academy of Dermatology Association to find a specialist.
Multidisciplinary Care Centers

Is It Time to Get a Second Opinion or Specialized Evaluation?
If you've visited your PCP, met with specialists, and undergone the recommended tests, but are still searching for a diagnosis, it may be time to visit an academic medical center or, for pediatric patients, a children's hospital. Academic medical centers and children's hospitals, often called multidisciplinary care centers, typically bring together specialists from different fields to work together on complex cases like rare diseases.
Multidisciplinary care centers may offer more coordinated care and be involved in clinical research, which may help reduce the time to diagnosis and provide access to emerging diagnostic tools. Specialists at these centers may have a deeper understanding of rare diseases and serve as a resource when you'd like a second opinion, particularly when test results or treatment plans are not delivering expected results.
Find hospitals that may partner with medical schools and programs in your area.
Children’s hospitals and large teaching hospitals may also offer dedicated specialists and programs for pediatric patients with undiagnosed or rare diseases. These programs bring pediatric experts together in one place and may provide more coordinated care for your child.
Search for children's or university hospitals in your area.
Rare Disease Experts

How can you find a rare disease expert?
If a diagnosis, care management, or treatment plan remains unclear despite extensive efforts by your PCP and specialists, it may be time to find a rare disease expert for your disease, if available. A rare disease expert is a medical provider that has knowledge or training on specific rare disease(s), but there may only be a few experts in your state, region, or country. Rare disease experts may work at large research or teaching hospitals, sometimes called centers of excellence. Centers of Excellence commit to sharing knowledge and best practices that can lead to improved care and treatment for individuals living with a rare disease.
You can also contact a GARD Information Specialist for help finding experts, centers of excellence, or clinics that focus on your disease.
Find Your Community
Patient organizations can help patients and families connect. They build public awareness of the disease and are a driving force behind research to improve patients' lives. They may offer online and in-person resources to help people live well with their disease. Many collaborate with medical experts and researchers.
Services of patient organizations differ, but may include:
- Ways to connect to others and share personal stories
- Easy-to-read information
- Up-to-date treatment and research information
- Patient registries
- Lists of specialists or specialty centers
- Financial aid and travel resources
Please note: GARD provides organizations for informational purposes only and not as an endorsement of their services. Contact a GARD Information Specialist for more information on organizations that may be dedicated to this disease. Please contact an organization directly if you have questions about the information or resources it provides.
View GARD's criteria for including patient organizations, which can be found under the FAQs on our About GARD page. Request an update or to have your organization added to GARD.

Patient Organizations
6 Organizations
Ichthyosis
United States
Rare Diseases
United States
Participate in Research
Clinical studies are a part of clinical research and play an important role in medical advances for rare diseases. Through clinical studies, researchers may ultimately uncover better ways to treat, prevent, diagnose, and understand human diseases.

What Are Clinical Studies?
- Clinical trials determine if a new test or treatment for a disease is effective and safe by comparing groups receiving different tests/treatments.
- Observational studies involve recording changes over time among a specific group of people in their natural settings.
Learn more about clinical trials from this National Institutes of Health webpage.
Why Participate in Clinical Studies?
To find the right clinical study we recommend you consult your doctors, other trusted medical professionals, and patient organizations. Additionally, you can use ClinicalTrials.gov to search for clinical studies by disease, terms, or location.
What if There Are No Available Clinical Studies?
ResearchMatch helps connect people interested in research studies with researchers from top medical centers across the United States. Anyone from the U.S. can register with this free program funded by NIH. Researchers from participating institutions use the database to search for and invite patients or healthy volunteers who meet their study criteria to participate.
Why may you want to consider joining the All of Us Research Program?
The All of Us Research Program is inviting 1 million people from all backgrounds across the U.S. to help build one of the most diverse health databases in history. Researchers will use the data to learn how our biology, lifestyle, and environment affect health. This may one day help them find ways to treat and prevent diseases.
Information Center
GARD collects data from a variety of sources to populate its website and provide accurate and reliable information on rare diseases.
GARD uses data collected from Orphanet, Online Mendelian Inheritance in Man (OMIM) , and Mondo Disease Ontology to interpret and provide information on rare diseases. This includes names, synonyms, genes, symptom frequency, population estimates and more.
- Orphanet is an online database of rare diseases and orphan drugs that provides aggregated data coordinated by INSERM-US14 in Paris.
- OMIM is a database of human genes and genetic phenotypes authored and edited at the McKusick-Nathans Institute of Genetic Medicine , Johns Hopkins University School of Medicine.
- Mondo Disease Ontology provides a logic-based structure unifying multiple disease resources in coordination with the Human Phenotype Ontology (HPO) and support from the NIH National Human Genome Research Institute Phenomics First Resource.
GARD uses the Human Phenotype Ontology (HPO) for standard terminology to represent a disease's phenotypic and clinical features.
GARD uses information gathered from the National Center for Biotechnology Information's MedGen to help in explaining genetic and rare diseases.
GARD uses the National Library of Medicine for a variety of resources on health information.
- Learn about the Newborn Screening Coding and Terminology Guide
- Find health information from MedlinePlus
- Learn about the Unified Medical Language System
GARD uses additional resources when developing content.
Last Updated: June 2026